Giorgio Olivo§¶, Martina Nardi§, Diego Vìdal¶, Alessia Barbieri§, Andrea Lapi§‡, Laura Gómez¶⊥, Osvaldo Lanzalunga§‡, Miquel Costas*¶, and Stefano Di Stefano*§‡
§ Università degli Studi di Roma, Rome, Italy
¶ Institut de Química Computacional i Catàlisi (IQCC), Girona, Spain
Inorg. Chem., 2015, 54 (21), pp 10141–10152
DOI: 10.1021/acs.inorgchem.5b01500
Publication Date (Web): October 12, 2015
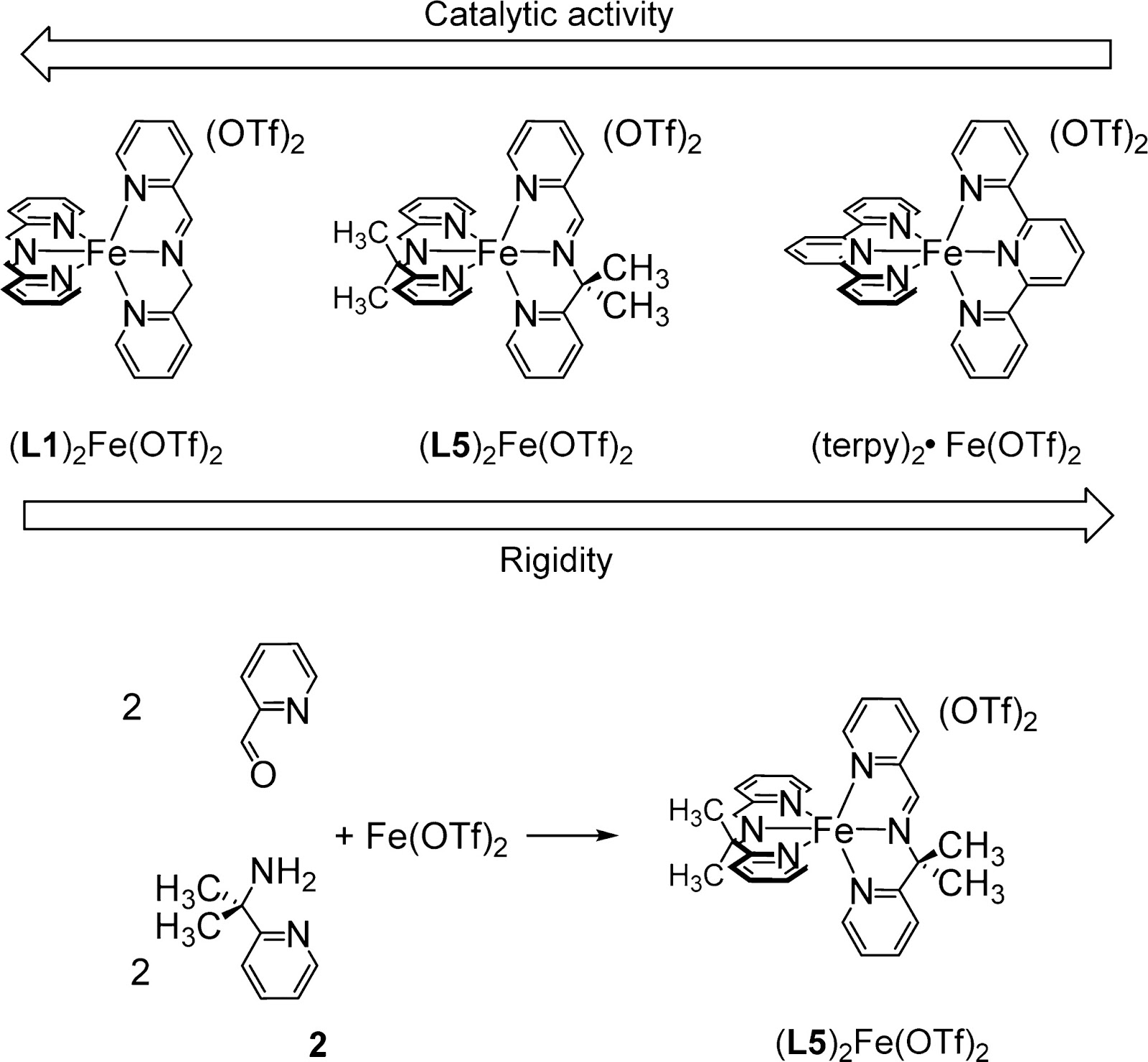
A family of imine-based nonheme iron(II) complexes (LX)2Fe(OTf)2 has been prepared, characterized, and employed as C–H oxidation catalysts. Ligands LX (X = 1, 2, 3, and 4) stand for tridentate imine ligands resulting from spontaneous condensation of 2-pycolyl-amine and 4-substituted-2-picolyl aldehydes. Fast and quantitative formation of the complex occurs just upon mixing aldehyde, amine, and Fe(OTf)2 in a 2:2:1 ratio in acetonitrile solution. The solid-state structures of (L1)2Fe(OTf)(ClO4) and (L3)2Fe(OTf)2 are reported, showing a low-spin octahedral iron center, with the ligands arranged in a meridional fashion. 1H NMR analyses indicate that the solid-state structure and spin state is retained in solution. These analyses also show the presence of an amine-imine tautomeric equilibrium. (LX)2Fe(OTf)2 efficiently catalyze the oxidation of alkyl C–H bonds employing H2O2 as a terminal oxidant. Manipulation of the electronic properties of the imine ligand has only a minor impact on efficiency and selectivity of the oxidative process. A mechanistic study is presented, providing evidence that C–H oxidations are metal-based. Reactions occur with stereoretention at the hydroxylated carbon and selectively at tertiary over secondary C–H bonds. Isotopic labeling analyses show that H2O2 is the dominant origin of the oxygen atoms inserted in the oxygenated product. Experimental evidence is provided that reactions involve initial oxidation of the complexes to the ferric state, and it is proposed that a ligand arm dissociates to enable hydrogen peroxide binding and activation. Selectivity patterns and isotopic labeling studies strongly suggest that activation of hydrogen peroxide occurs by heterolytic O–O cleavage, without the assistance of a cis-binding water or alkyl carboxylic acid. The sum of these observations provides sound evidence that controlled activation of H2O2 at (LX)2Fe(OTf)2 differs from that occurring in biomimetic iron catalysts described to date.


§ Università degli Studi di Roma, Rome, Italy
¶ Institut de Química Computacional i Catàlisi (IQCC), Girona, Spain
Inorg. Chem., 2015, 54 (21), pp 10141–10152
DOI: 10.1021/acs.inorgchem.5b01500
Publication Date (Web): October 12, 2015
A family of imine-based nonheme iron(II) complexes (LX)2Fe(OTf)2 has been prepared, characterized, and employed as C–H oxidation catalysts. Ligands LX (X = 1, 2, 3, and 4) stand for tridentate imine ligands resulting from spontaneous condensation of 2-pycolyl-amine and 4-substituted-2-picolyl aldehydes. Fast and quantitative formation of the complex occurs just upon mixing aldehyde, amine, and Fe(OTf)2 in a 2:2:1 ratio in acetonitrile solution. The solid-state structures of (L1)2Fe(OTf)(ClO4) and (L3)2Fe(OTf)2 are reported, showing a low-spin octahedral iron center, with the ligands arranged in a meridional fashion. 1H NMR analyses indicate that the solid-state structure and spin state is retained in solution. These analyses also show the presence of an amine-imine tautomeric equilibrium. (LX)2Fe(OTf)2 efficiently catalyze the oxidation of alkyl C–H bonds employing H2O2 as a terminal oxidant. Manipulation of the electronic properties of the imine ligand has only a minor impact on efficiency and selectivity of the oxidative process. A mechanistic study is presented, providing evidence that C–H oxidations are metal-based. Reactions occur with stereoretention at the hydroxylated carbon and selectively at tertiary over secondary C–H bonds. Isotopic labeling analyses show that H2O2 is the dominant origin of the oxygen atoms inserted in the oxygenated product. Experimental evidence is provided that reactions involve initial oxidation of the complexes to the ferric state, and it is proposed that a ligand arm dissociates to enable hydrogen peroxide binding and activation. Selectivity patterns and isotopic labeling studies strongly suggest that activation of hydrogen peroxide occurs by heterolytic O–O cleavage, without the assistance of a cis-binding water or alkyl carboxylic acid. The sum of these observations provides sound evidence that controlled activation of H2O2 at (LX)2Fe(OTf)2 differs from that occurring in biomimetic iron catalysts described to date.

コメント